CASE STUDY
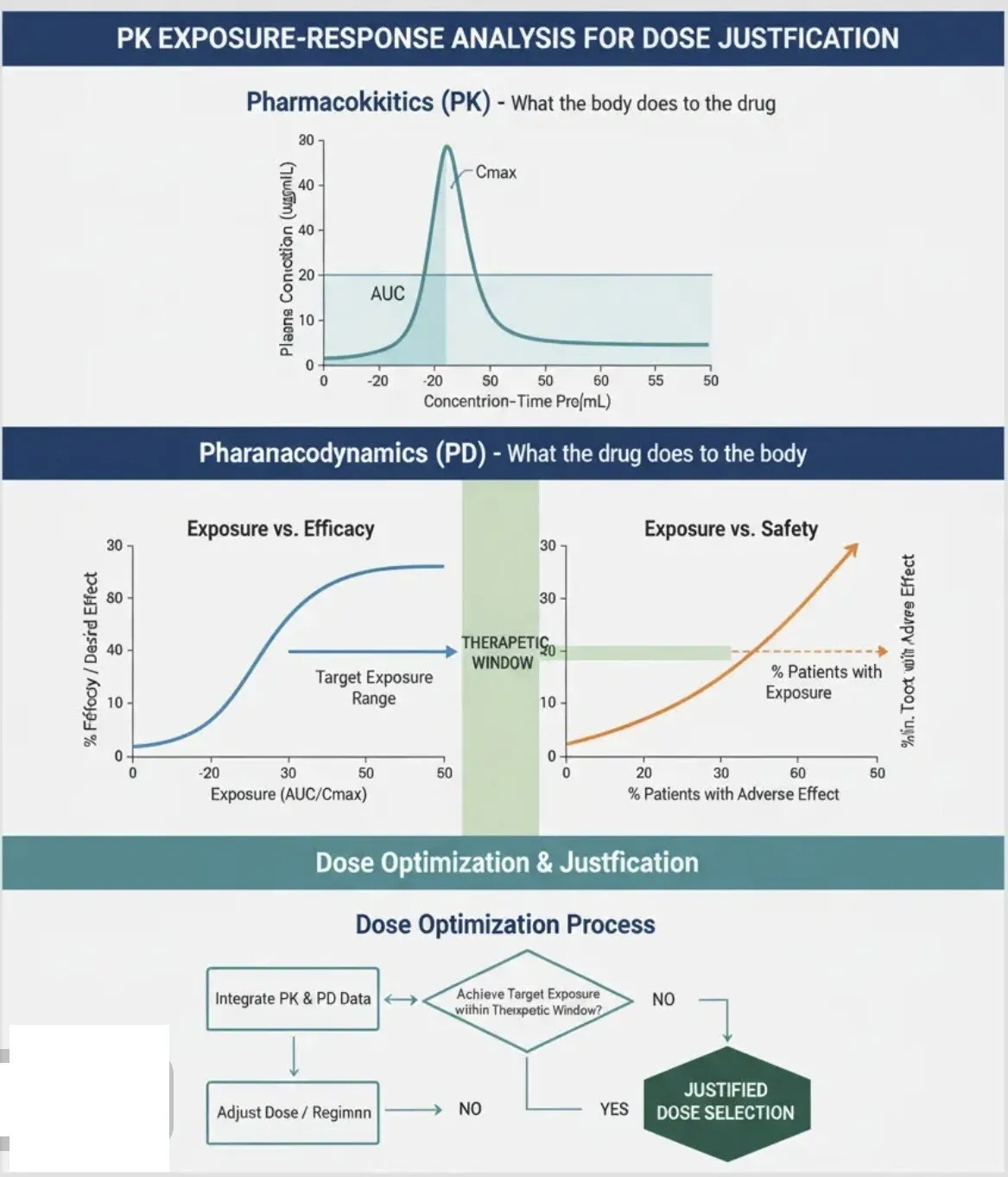
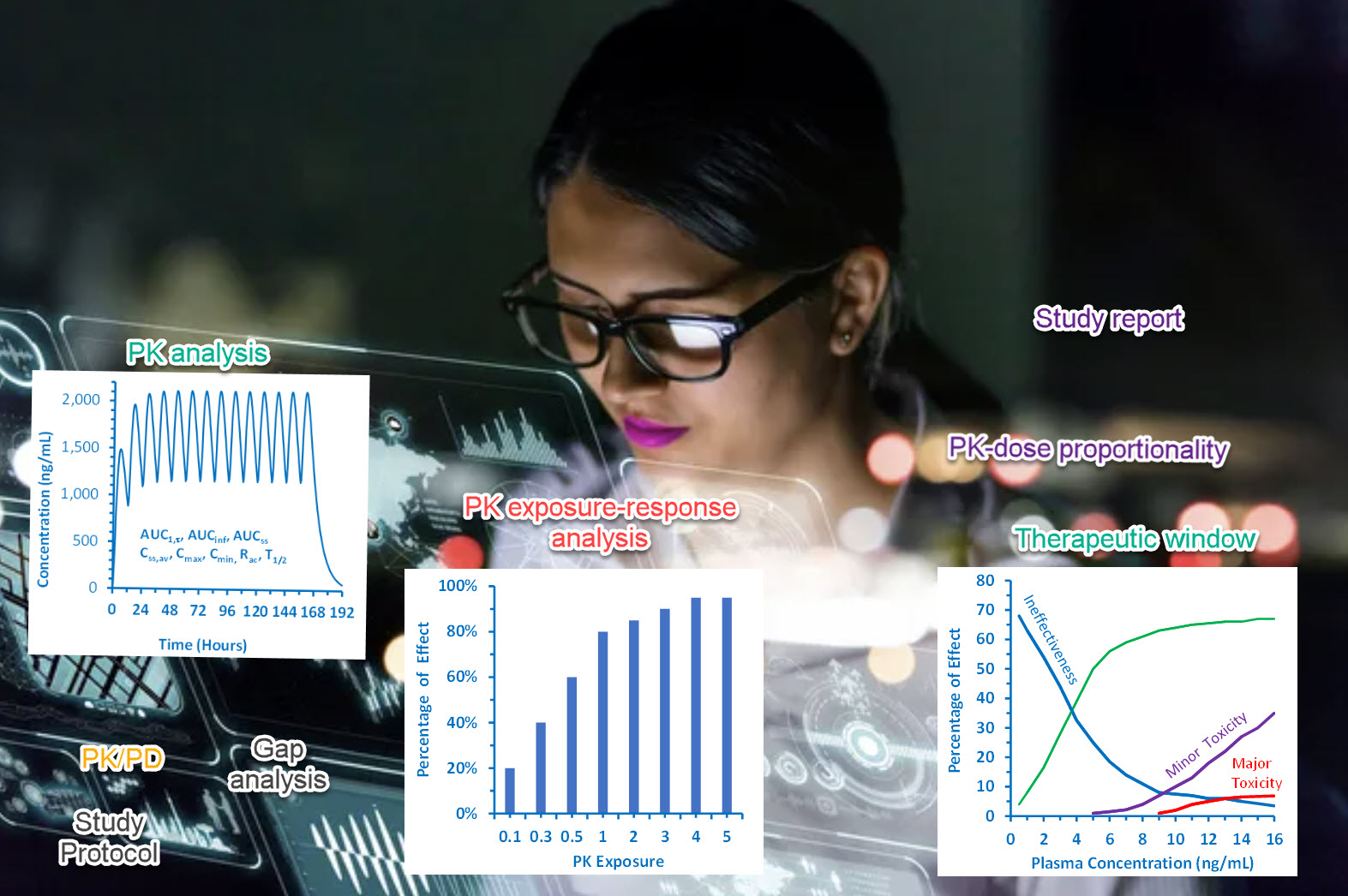
Dose Justification with PK Exposure-Response Analysis
Assessment of Drug-Drug Interaction Potential
Determine First-in-Human Study Doses with M&S
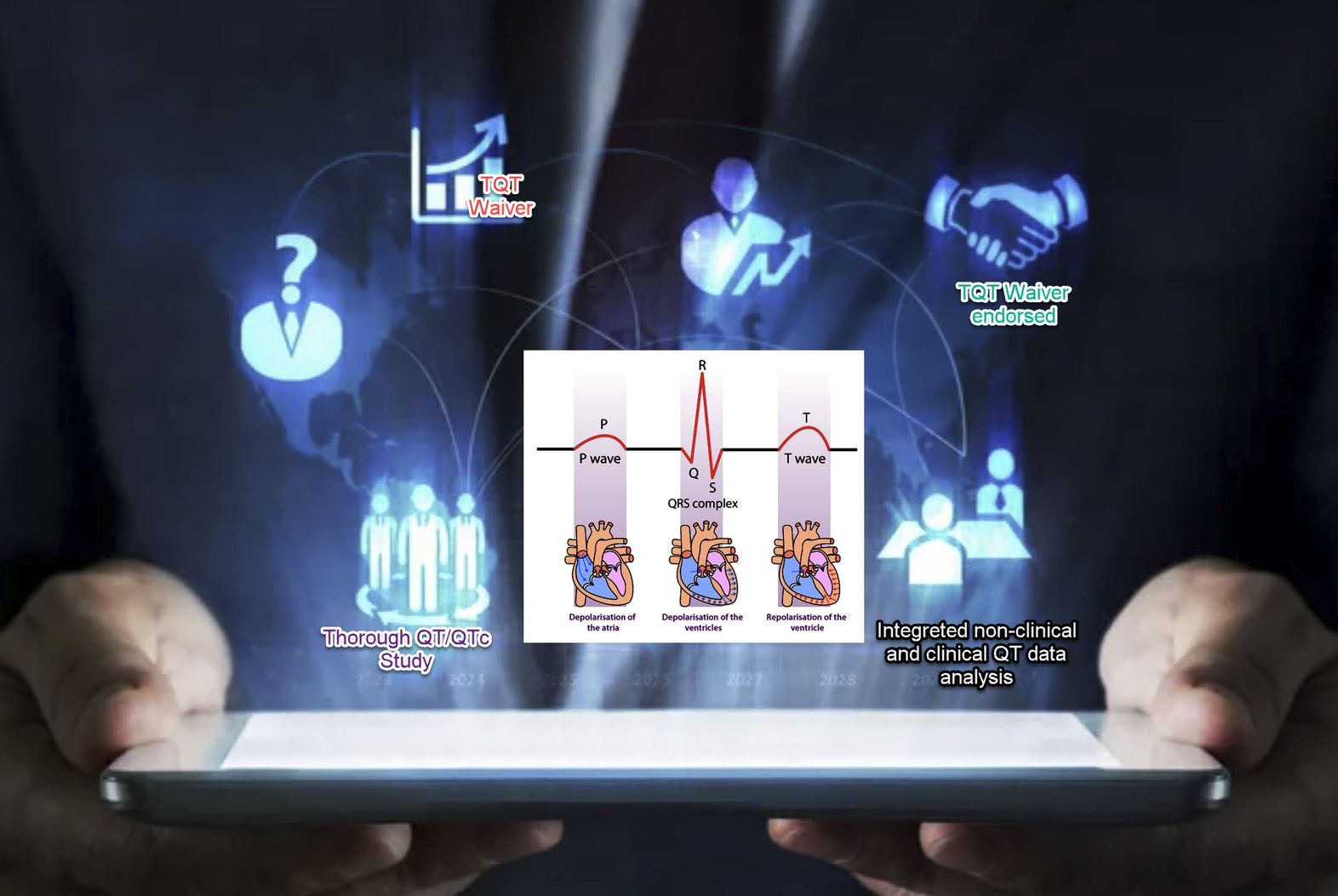
Risk Assessment for QT Prolongation Potential
New York, NY 10011
©Copyright 2019 to 2026. All Rights Reserved